 Riaan Research Initiative is seeking a clinical partner to conduct a small and short study involving analyzing serum samples from children with Cockayne Syndrome. The study’s goal is to assess GDF-15 serum levels in a small number of children with Cockayne Syndrome and determine whether the levels are elevated, and if so, how elevated. This study is based on a recent paper that hypothesizes that GDF-15, a growth differentiation factor that tells the brain to reduce appetite and induces weight loss, may be perpetually released in Cockayne Syndrome children, causing adverse effects.
Researchers from Dr. KJ Patel’s lab discovered that when formaldehyde is introduced to the body, it causes DNA damage. The kidneys subsequently release GDF-15, which triggers appetite suppression while the body attempts to repair the DNA damage. This response is also commonly seen in patients undergoing chemotherapy. Scientists believe that the body may have developed this evolutionary appetite block to prevent a person from ingesting more toxins while it is repairing DNA damage. Researchers theorize that as DNA damage builds up in Cockayne Syndrome patients, GDF-15 continues to be released because the DNA damage is never repaired, as CS children lack CSA or CSB, the DNA repair genes. This may explain one of the most prevalent phenotypes in Cockayne Syndrome: a failure to thrive and cachexia.
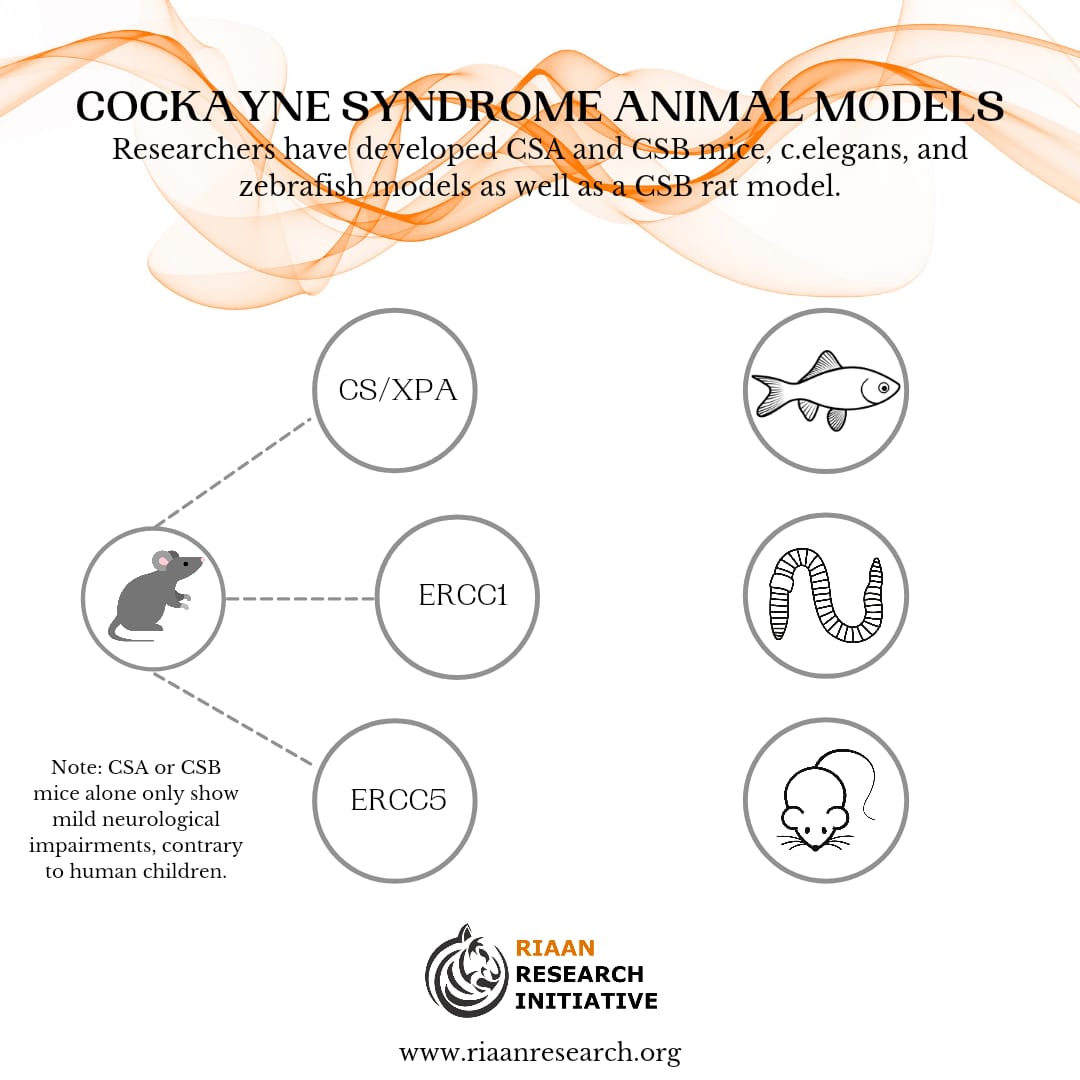
The authors tested this theory on an ADH5-CSB mice model. A CSB mice model alone only has mild neurological symptoms of Cockayne Syndrome, but once the aldehyde (ADH5) was introduced, and GDF-15 began to be released, the mice developed severe symptoms of the disease. However, the good news was that an antibody to GDF-15, developed by Pfizer, was able to alleviate the symptoms in mice. Our study seeks to test this phenomenon in children, and determine, at a minimum, whether the GDF-15 levels are consistently elevated, and whether the Pfizer antibody to GDF-15 could potentially be safe and effective for children. All testing will be conducted at Mayo Clinic Laboratories.
For more information or if you are interested in working with us on this project, please contact us at info@riaanresearch.org.
Riaan Research Initiative is seeking a clinical partner to conduct a small and short study involving analyzing serum samples from children with Cockayne Syndrome. The study’s goal is to assess GDF-15 serum levels in a small number of children with Cockayne Syndrome and determine whether the levels are elevated, and if so, how elevated. This study is based on a recent paper that hypothesizes that GDF-15, a growth differentiation factor that tells the brain to reduce appetite and induces weight loss, may be perpetually released in Cockayne Syndrome children, causing adverse effects.
Researchers from Dr. KJ Patel’s lab discovered that when formaldehyde is introduced to the body, it causes DNA damage. The kidneys subsequently release GDF-15, which triggers appetite suppression while the body attempts to repair the DNA damage. This response is also commonly seen in patients undergoing chemotherapy. Scientists believe that the body may have developed this evolutionary appetite block to prevent a person from ingesting more toxins while it is repairing DNA damage. Researchers theorize that as DNA damage builds up in Cockayne Syndrome patients, GDF-15 continues to be released because the DNA damage is never repaired, as CS children lack CSA or CSB, the DNA repair genes. This may explain one of the most prevalent phenotypes in Cockayne Syndrome: a failure to thrive and cachexia.
The authors tested this theory on an ADH5-CSB mice model. A CSB mice model alone only has mild neurological symptoms of Cockayne Syndrome, but once the aldehyde (ADH5) was introduced, and GDF-15 began to be released, the mice developed severe symptoms of the disease. However, the good news was that an antibody to GDF-15, developed by Pfizer, was able to alleviate the symptoms in mice. Our study seeks to test this phenomenon in children, and determine, at a minimum, whether the GDF-15 levels are consistently elevated, and whether the Pfizer antibody to GDF-15 could potentially be safe and effective for children. All testing will be conducted at Mayo Clinic Laboratories.
For more information or if you are interested in working with us on this project, please contact us at info@riaanresearch.org.